![]()
Les Assises de Génétique Humaine et Médicale sont une conférence annuelle qui rassemble des experts en génétique humaine et médicale pour discuter des dernières avancées dans le domaine.
La 12ème édition de cette conférence s’est tenue du 9 au 12 janvier 2024 au Palais des Congrès de Paris . Les thèmes abordés lors de cette conférence incluent les traitements innovants géniques ou dérivés de la génétique, la génétique du développement et sa modélisation animale ou cellulaire, la lecture évolutionniste de la génétique humaine, la génétique des maladies infectieuses, à l’honneur dans ces années de pandémie, ou encore l’analyse de nos pratiques, notamment en matière d’accès à la médecine génomique personnalisée.
Le centre de référence 2M2C était présent à cet évènement avec plusieurs posters :
-
Impact de la perte de fonction de PRDM13 dans le développement du cervelet et du tronc cérébral.
Les pathologies congénitales du cervelet forment un groupe hétérogène de maladies neurodéveloppementales, affectant 1 enfant sur 7000 naissances. Le séquençage à haut débit a permis d’identifier nombre de variants génétiques, mais ~50% des patients restent sans diagnostic concluant. De manière surprenante, peu de mutations ont été caractérisées dans des gènes codant pour des facteurs de transcription clés du développement neural. Récemment, nous avons identifié des mutations bialléliques dans PRDM13 chez des enfants atteints dès la naissance d’hypotonie, de troubles des fonctions autonomes (déglutition, respiration), et d’absence de développement moteur et cognitif. Les examens fœtopathologiques et par neuroimagerie révèlent une hypoplasie sévère et précoce du cervelet, associée à une réduction et une désorganisation de la couche de cellules de Purkinje, ainsi qu’une atteinte des noyaux de l’olive inférieure (NOI). PRDM13 code pour un répresseur transcriptionnel, connu comme un acteur clé dans la spécification neurale dans la rétine et la moelle épinière. Néanmoins, ses fonctions dans le développement du cerveau postérieur et plus spécifiquement du cervelet n’avaient pas encore été décrites. Afin de valider l’implication des mutations de PRDM13 dans la pathologie, et comprendre le mécanisme sous-jacent, nous avons analysé l’expression et le rôle de prdm13 chez le poisson-zèbre. Nous avons ainsi démontré que prdm13 est exprimé dans des progéniteurs spécifiques du cervelet et du cerveau postérieur, de façon similaire aux observations faites sur des échantillons humains embryonnaires. De plus, des expériences de lignage cellulaire montrent que, dans les rhombomères postérieurs, les progéniteurs exprimant prdm13 génèrent de nombreuses populations neuronales, activatrices et inhibitrices, localisées au niveau dorsal, ainsi que les NOI. Dans le cervelet, leur destin est plus restreint ; ils donnent naissance à une sous-population de cellules de Purkinje et à quelques interneurones GABAergiques. Chez les poissons-zèbre mutants pour prdm13, un défaut de spécification dans les rhombomères postérieurs conduit à l’absence totale de NOI ainsi que de certaines populations neuronales de la colonne viscero-sensitive. Dans le cervelet, le phénotype est moins marqué avec une réduction modérée du nombre de certaines cellules de Purkinje. Nos travaux mettent en évidence l’effet pathogénique des variants de PRDM13 et permettent de comprendre les caractéristiques cliniques observées chez les enfants atteints. Nos données révèlent également l’impact différentiel de la perte de fonction de prdm13 entre les différents rhombomères. Pour décrypter les mécanismes sous-jacents, nous étudions actuellement l’impact de la perte de fonction de prdm13 sur le transcriptome des progéniteurs prdm13. En parallèle, nous mettons au point un modèle organoïde humain de cervelet muté pour PRDM13, pour élucider son rôle dans la formation du cervelet.
-
Retour d’expérience sur la mise en place du séquençage de l’ARN messager (mRNA-seq) dans un laboratoire de neurogénétique moléculaire en diagnostic de routine.
Le séquençage à haut débit de l’ARN (RNA-seq) constitue une approche de validation fonctionnelle complémentaire à celle du séquençage de l’ADN. Il permet la mise en évidence de jonctions aberrantes suite à un épissage alternatif ou à la présence de gènes de fusion, la détection de variants mais aussi l’expression différentielle des gènes. Plusieurs études ont montré l’intérêt majeur de cette technique qui améliore le rendement diagnostique et permet de reclassifier des variations de signification incertaine représentant un challenge pour les laboratoires de génétique en l’absence de données d’études fonctionnelles.
Nous avons mis en place dans notre laboratoire de neurogénétique moléculaire une double approche qualitative du séquençage de l’ARN messager ciblé et total dans un contexte de caractérisation de variants de signification incertaine ayant un impact prédit sur l’épissage et mis en évidence préalablement en DNA-seq panel, exome et aussi en Génome (plateformes nationales SeqOIA et AURAGEN). Les ARNs sont extraits à partir de cellules lymphocytaires (Tube Paxgene) ou de fibroblastes (Biopsie de peau), et plus rarement à partir de tissu fœtal (Muscle, Poumon) selon l’expression du gène d’intérêt. Nous procédons par la suite à la sélection des ARNm par la capture de la queue polyA puis déplétion ou non en globine et en ARN ribosomal selon l’analyse souhaitée et le type d’échantillon utilisé. Pour l’analyse mRNA ciblée, nous capturons par la suite 250 gènes impliqués dans nos pathologies d’intérêt (maladies congénitales ou très précoces du cervelet et du tronc cérébral, et mouvements anormaux à début pédiatrique). Le traitement bioinformatique des données a été initialement assuré par Génosplice lors de la mise en place de la technique, puis nous avons effectué une transition vers une utilisation en routine diagnostique sur la plateforme MOABI(APHP), Ces deux pipelines intègrent l’outil d’alignement STAR pour l’analyse qualitative des jonctions aberrantes. L’interprétation des résultats est réalisée d’une part à partir d’un fichier de comptage de jonctions présentes au niveau de l’échantillon et le calcul du ratio des jonctions d’inclusion et d’exclusion et d’autre part par la visualisation des BAMs jonctions en Sashimi Plot sur IGV. 82% des gènes panel sont analysables à partir d’ARN extrait sur Tube Paxgene contre 63 % des gènes mRNA total.
Cette approche nous a permis de reclasser à ce jour 57% des variants testés de signification incertaine en probablement pathogène ou pathogène selon la classification ACMG et confirme l’intérêt de l’implémentation de cette technique en routine dans un laboratoire de diagnostic. Plusieurs exemples de RNAseq ciblé et total seront présentés ainsi qu’un comparatif des différents processus techniques utilisés.
-
A la recherche de la mutation perdue : Pensez aux insertions d’éléments mobiles !

Les rétrotransposons sont des segments d’ADN qui se déplacent à travers le génome par un mécanisme de « copier-coller » en utilisant l’ARN comme intermédiaire. Chez l’humain, les rétrotransposons représentent jusqu’à environ la moitié du génome. Certains d’entre eux, les LINE1, Alu et SVA sont toujours actifs et peuvent générer de nouvelles copies connues sous le nom d’insertions d’éléments mobiles (MEIs). Ils peuvent ainsi être à l’origine de maladies rares, soit en interrompant la séquence codante, soit en altérant l’ARN messager. Nous avons mis en évidence la présence d’un de ces éléments mobiles comme second hit chez trois patients avec un phénotype spécifique d’une pathologie récessive et porteur d’une seule mutation hétérozygote dans le gène d’intérêt.
Le patient 1 présentait un syndrome de Joubert typique clinique et radiologique ; le patient 2, un tableau typique de syndrome de Poretti-Boltshauser avec des kystes cérébelleux caractéristiques, évoquant l’implication du gène LAMA1 ; La patiente 3, une encéphalopathie avec une leucodystrophie à l’IRM, des anomalies de la spectroscopie et un bilan métabolique caractéristiques d’une maladie de Canavan, orientant vers l’étude spécifique du gène ASPA.
Les deux premiers patients ont bénéficié d’une analyse moléculaire par séquençage à haut débit d’un panel de gènes impliqués dans les maladies congénitales ou très précoces du cervelet et du tronc cérébral. Un variant hétérozygote hérité de la mère de type stop dans le gène CC2D2A a été mis en évidence chez le patient 1, et une délétion hétérozygote paternelle de 12 exons a été détectée dans le gène LAMA1 chez le patient 2. Le patient 3 a quant à lui bénéficié d’une analyse ciblée du gène ASPA qui a montré la présence d’un faux-sens hétérozygote, connu pathogène dans la littérature, hérité de la mère. Ces 3 patients ont ensuite eu une analyse de Génome en trio (AURAGEN pour le patient 1 et SeqOIA pour les deux autres) à la recherche d’un second variant éventuellement intronique, mais l’analyse a été non conclusive. Vu la spécificité clinique, une recherche manuelle à partir des données d’alignement a permis la mise en évidence d’une insertion d’élément mobile de type Line1 et Alu chez ces patients respectivement au niveau d’un exon, d’une jonction intron-exon, et d’un intron, héritée du parent ne transmettant pas le variant précédemment identifié.
L’insertion d’élément mobile est un mécanisme responsable de maladies rares certainement sous-estimé et ceci est due à l’absence d’algorithmes bioinformatiques performants capables de détecter ces évènements à partir des données de séquençage à haut débit de court fragment, utilisé à ce jour en routine diagnostique.
-
« tubulinopathies » : description clinico-radiologique de 23 patients et revue de la littérature.
Les tubulinopathies définissent un groupe de malformations cérébrales en lien avec des variants pathogènes dans un des gènes codant pour différents isotypes de tubuline. Cliniquement, la plupart des patients présentent un retard global de développement, une déficience intellectuelle modérée à sévère, une épilepsie et des troubles moteurs. L’IRM cérébrale montre classiquement une anomalie de fragmentation des noyaux gris centraux, associée fréquemment à des anomalies de la giration (polymicrogyrie, lissencéphalie), du corps calleux ou de la fosse postérieure.
La plupart des mutations sont de novo, mais quelques familles ont été décrites dans la littérature, avec une transmission dominante, avec un phénotype décrit parfois moins sévère. Des cas de formes « mild » radiologiques, c’est à dire sans anomalie de giration ont également été décrits chez des fœtus, hérités pour certains de parents pauci ou asymptomatique. L’objectif principal de cette étude est de décrire un nouveau phénotype clinico-radiologique de patients avec une forme « mild » de tubulinopathie.
Nous avons donc inclus, à partir d’un appel à collaboration européen, les adultes ou enfants de plus de 6 ans, porteurs d’un variant pathogène dans TUBA1A, TUBB2B, TUBB3, TUBB ou TUBB2A avec une cognition normale ou une déficience intellectuelle légère (QIT >70). Les données cliniques, neuropsychologiques et d’IRM cérébrales ont été recueillies.
23 patients issus de 15 familles non apparentées ont été inclus. Le gène majoritairement retrouvé est TUBB3 avec 52% de patients porteurs. 6 variants sont de novo et 8 hérités.
78% ont présenté un retard psychomoteur, seulement 16% ont présenté une épilepsie, et 88% présentent des troubles des apprentissages. Aucun des patients ne présentent d’anomalie de giration à l’IRM cérébrale.
La corrélation génotype – phénotype a permis de mettre en évidence deux variants récurrents (TUBB3 p. Pro357Leu et TUBB p.Asn52Ser), à la fois dans notre étude et dans la littérature, chez des patients présentant le même phénotype. Toutefois, pour 4 variants, il existe une variabilité phénotypique intra et inter familiale.
Notre étude confirme l’existence d’un phénotype « mild » de tubulinopathie, avec, pour certains variants, une corrélation génotype phénotype, importante pour l’information pronostique.
-
Phénotype neurodéveloppemental et cérébelleux des patients avec mutation gain de fonction du canal TRPM3.
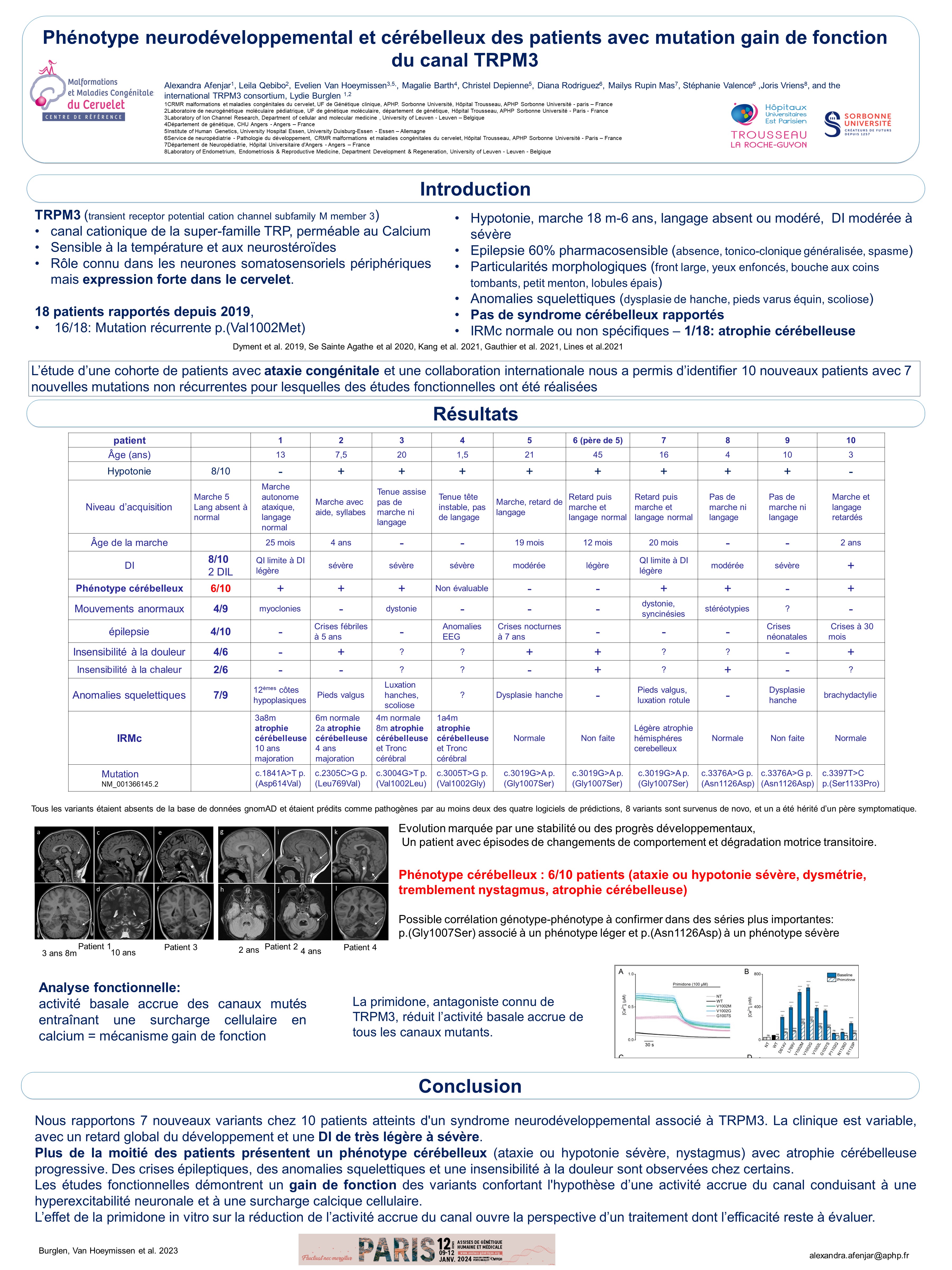
TRPM3 est un canal cations localisé à la membrane plasmatique, sensible à la température et aux neurostéroïdes, très exprimé dans le cervelet, et important pour l’homéostasie du calcium. Des variants de novo de TRPM3, dont une mutation majoritaire, ont été identifiés chez des patients avec trouble du neurodéveloppement (retard psychomoteur, hypotonie, épilepsie, déficience intellectuelle, diminution de la sensibilité au chaud et à la douleur). Lors de l’exploration génétique des patients du CRMR atteints d’ataxie congénitale, nous avons identifié 4 nouveaux patients présentant un phénotype cérébelleux associé à un variant de novo de TRPM3. Nous décrivons le phénotype de 10 patients (8 filles, 2 garçons; 21 mois-45 ans) recrutés par une collaboration internationale, porteurs de 7 mutations de TRPM3 distinctes. Ces patients présentent un large éventail de symptômes, en terme de retard moteur, déficience intellectuelle (sévère 4/10, légère 3/10), épilepsie (2/10 et crises fébriles 1/10), anomalies squelettiques (7/10: subluxation de hanche, dislocation rotulienne, maladie de Perthes, brachydactylie, pieds valgus, anomalie des côtes). Deux sur 7 ont un retard statural et 3/10 une microcéphalie postnatale modérée. Le phénotype cérébelleux, ataxie ou hypotonie sévère, nystagmus, atrophie cérébelleuse, est objectivé chez 6/10 patients. Une collaboration avec l’équipe de Joris Vriens à Leuven a permis de confirmer le mécanisme gain de fonction pour tous les variants, caractérisé par une activité basale accrue entraînant une surcharge cellulaire en calcium. La primidone, antiépileptique antagoniste connu du TRPM3, réduit l’activité basale accrue de tous les canaux mutants.
Ce travail confirme l’existence d’un spectre de troubles du neurodéveloppement autosomique dominant avec une composante cérébelleuse fréquente, lié à un gain de fonction de TRPM3, et incite à l’évaluation des antagonistes du TRPM3 comme thérapie potentielle.