Qu’est-ce que le syndrome de Joubert ?
C’est une maladie rare, d’origine génétique, en rapport avec une malformation du cervelet et du tronc cérébral.
Quelles sont les signes ?
Chez le nourrisson, on observe une hypotonie, parfois des troubles du rythme respiratoire, et des mouvements anormaux des yeux.
Chez l’enfant plus grand, on observe des troubles de l’équilibre (ataxie) et de la coordination des mouvements, des troubles de l’oculomotricité, des difficultés d’apprentissage.
Certains enfants présentent également une atteinte hépatique, rénale et/ou rétinienne.
Comment fait-on le diagnostic ?
Le diagnostic est évoqué à partir des signes cliniques que présente l’enfant et il est confirmé par l’Imagerie par Résonance Magnétique (IRM) : petit vermis et apparence d’une dent molaire.
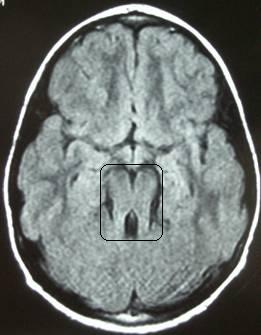 Signe de la dent molaire à l’IRM
Signe de la dent molaire à l’IRM
Prévalence et évolution
La prévalence du syndrome de Joubert est estimée à 1 enfant pour 100 000 et il concerne aussi bien les filles que les garçons. L’évolution est variable d’un enfant à l’autre, chaque enfant progresse à son rythme.
Quel est le mode de transmission ?
Nous avons tous de nombreux gènes et nous possédons deux copies de chaque gène, une provenant de notre père et une de notre mère. Le syndrome de Joubert se transmet, dans la plupart des cas (à une exception près), sur un mode autosomique récessif, ce qui signifie que le syndrome s’exprime et l’enfant est atteint lorsque les 2 copies du gène présentes chez cet enfant, celle venant du père et celle venant de la mère, sont altérées/mutées.
Les parents d’un enfant atteint du syndrome de Joubert sont tous les deux porteurs d’une copie altérée du gène et d’une copie non altérée ; on dit qu’ils sont hétérozygotes, le syndrome ne s’exprime pas chez eux.
A chaque naissance, un couple qui a un enfant atteint du syndrome de Joubert, a une probabilité de 25% d’avoir un enfant malade et de 75% d’avoir un enfant indemne.
Quel suivi médical pour mon enfant ?
Le suivi des enfants atteints d’un syndrome de Joubert peut être assuré par un neuropédiatre ou un généticien. Il est important pour évaluer le développement psychomoteur et proposer, lorsque c’est nécessaire, une rééducation adaptée mais aussi pour surveiller le bilan rénal et hépatique (environ une fois par an ou tous les 2 ans). Parfois un suivi par un néphrologue (spécialiste du rein) ou un hépatologue (spécialiste du foie) est nécessaire. Très souvent, les enfants atteints ont besoin d’un suivi par un ophtalmologue et/ou un orthoptiste.
Pourquoi me propose t-on de rencontrer un psychologue ?
Le moment de l’annonce du diagnostic et ses conséquences peut être délicat et douloureux, c’est pourquoi un accompagnement psychologique est proposé à l’enfant et à sa famille. Des bilans neuropsychologiques sont également proposés afin de suivre l’évolution des capacités cognitives de l’enfant, d’orienter chaque enfant vers des prises en charge adaptées, et de proposer des aménagements ou des orientations scolaires.
Pourquoi me propose t-on une consultation de génétique ?
Cette consultation permet de discuter du mode de transmission du syndrome de Joubert, du risque de récidive de cette malformation lors d’une nouvelle grossesse, et de la façon de suivre une nouvelle grossesse. Un prélèvement sanguin pourra être réalisé chez l’enfant et ses parents pour une étude génétique, qui peut être longue, car il existe au moins 20 gènes responsables de syndrome de Joubert.
Où en est la recherche ?
Le premier gène impliqué dans le syndrome de Joubert a été découvert en 2004. A ce jour 20 gènes ont été identifiés. Toutefois, les études se poursuivent pour identifier les gènes encore inconnus et mieux comprendre leur rôle. Le syndrome de Joubert est une ciliopathie, c’est-à-dire une maladie du cil primitif. Les cils primitifs sont de petits organites qui forment des extensions de la cellule. Ils sont composés de plusieurs centaines de protéines différentes. Les gènes impliqués dans le syndrome de Joubert codent pour certaines de ces protéines, mais on ne connaît pas encore les mécanismes qui conduisent à la malformation.